Priony określić można jako “białkowe czynniki zakaźne”. Wywołują one szereg śmiertelnych chorób określanych wspólnie jako przenośne encefalopatie gąbczaste (ang. transmissible spongiform encephalopathies, TSE). Chociaż pierwsza choroba prionowa została poznana już w XVIII wieku i dotyczyła ona owiec (choroba scrapie), to jej zakaźność udowodniono eksperymentalnie dopiero w 1939 roku [1].
Mimo zbliżonych efektów tkankowych – choroby neurodegeneracyjne na przykładzie choroby Parkinsona lub choroby Alzheimera znacznie różnią się od chorób prionowych. Schorzenia wywoływane przez priony posiadają pewną unikatową cechę – możliwa jest ich transmisja człowiek-człowiek, zwierzę-zwierzę i człowiek-zwierzę [2].
W przeciwieństwie do innych chorób infekcyjnych – najważniejszą cechą wyróżniającą choroby prionowe jest brak materiału genetycznego w czynniku etiologicznym. Postuluje się, że znaczna większość chorób prionowych wywoływana jest przez białko PrPSc o specyficznej konformacji, która, w przeciwieństwie do konformacji białka natywnego PrPC, jest bogata w regiony β-kartki. PrPC jest glikolizowanym białkiem transbłonowym, które ulega ekspresji przede wszystkim w komórkach ośrodkowego układu nerwowego (OUN), jednak można je znaleźć również w innych tkankach [3]. Fizjologiczna rola białka PrPC nie jest w pełni poznana, jednak obecnie uważa się, że białko to uczestniczy w procesach redukujących komórkowy stres oksydacyjny poprzez np. wiązanie jonów miedzi. Wśród innych funkcji białka PrPC wyróżnia się: uczestnictwo w procesach transdukcji sygnału, regulacja apoptozy, uczestnictwo w komunikacji synaptycznej poprzez stabilizację struktury synaps [4].
Komórkowe białko prionowe i jego formy
PrPC
Białko PrPC, od którego bezpośrednio pochodzi prionowy czynnik zakaźny, jest kodowane przez gen PRNP zlokalizowany na chromosomie 20. Ten konserwowany ewolucyjnie gen koduje polipeptyd o długości 253 aminokwasów. W białku tym wykazano również obecność dwóch terminalnych domen: (I) N-końcowy peptyd sygnałowy (1-22 aa); (II) hydrofobowa domena C-końcowa (232-253 aa). Po syntezie białko PrPC ulega licznym modyfikacjom post-translacyjnym w świetle siateczki śródplazmatycznej (ER): (I) usunięcie N- i C-końcowych peptydów sygnałowych; (II) glikozylacja; (III) utworzenie mostka disiarczkowego między resztami cysteiny w lokalizacjach Cys-179 i Cys-214; (IV) dodanie kotwicy glikozylofosfatydyloinozytolowej (GPI). W pełni dojrzały polipeptyd PrPC składa się więc z 209 aminokwasów i ulega prezentacji na błonie komórkowej. Prawidłowa trzeciorzędowa struktura białka składa się z trzech α-helis i dwóch antyrównoległych β-kartek w domenie C-końcowej, których zaburzenie jest kluczowe do przejścia PrPC → PrPSc. Region N-końcowy nie jest w pełni ustrukturyzowany i zawiera charakterystyczne czterokrotne powtórzenie ugrupowanie PHGGGWGQ, które uczestniczy w wiązaniu jonów miedzi (II) i kilku innych kationów dwuwartościowych metali przejściowych [5].
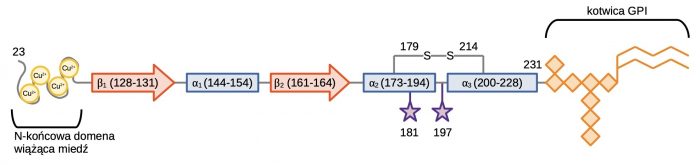
Dojrzały ludzki PrPC zbudowany jest z 209 aminokwasów. W jego skład wchodzą: (I) elastyczna domena N-końcowa zawierającej cztery powtórzenia PHGGGWGQ wiążące miedź; (II) złożona domena C-końcowej zawierająca trzy struktury α-helikalne i dwie β-kartki. Aminokwasy w pozycjach Cys-179 i Cys-214 tworzą wiązanie disiarczkowe między domenami α2 i α3. Dwa miejsca glikozylacji znajdują się przy resztach 181. i 197., a kotwica GPI jest połączona z terminalną resztą 231.
W komórce PrPC lokalizuje się głównie w błonach. Pod względem globalnym – białko to jest wykrywane przede wszystkim w OUN, jednak pewne jego formy udało się zaobserwować również w innych tkankach: sercu, mięśniach szkieletowych, tkankach limfatycznych, nerkach, śródbłonku, skórze i w przewodzie pokarmowym. Wśród komórek OUN PrPC zidentyfikowano w neuronach i komórkach glejowych. W warunkach fizjologicznych PrPC związane jest z błoną poprzez kotwicę GPI, a w przypadku występowania białka prionowego w cytozolu powiązano ten stan z neurotoksycznością (nawet w przypadku białka poprawnie pofałdowanego) [6].
PrPSc
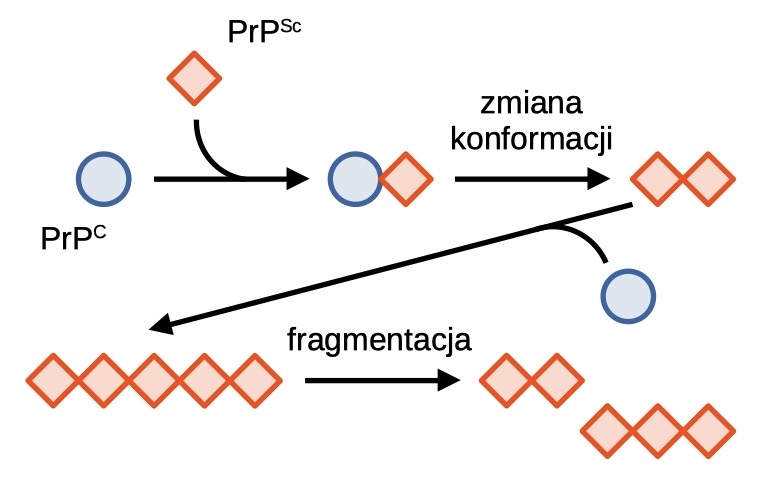
Ogólnie przyjmuje się, że głównym czynnikiem etiologicznym chorób prionowych jest nieprawidłowo pofałdowane białko PrP, które przyjmuje strukturę bogatą w struktury β-kartek. Tak zmieniona struktura białka jest niezwykle stabilna, oporna na proteazy i zdolna do tworzenia nierozpuszczalnych w wodzie agregatów. Takie agregaty polimeryzują w dłuższe włókna amyloidowe. Włókna te są kruche i mogą pękać dając nowe zarodki polimeryzacji [5]. W jaki sposób białko z formy PrPC przekształca się w patogenną postać PrPSc? Dokładny mechanizm konformacyjnej zmiany nie jest w pełni poznany, jednak wiadomo, że wspomagany jest on poprzez kontakt białka PrPC z białkiem PrPSc, w czasie którego białko patogenne działa jak matryca stymulująca przejście konformacyjne. Analizując ten proces, można zauważyć, że postęp choroby jest uwarunkowany obecnością pierwotnego białka prionowego, które zmienia formy fizjologiczne do postaci patogennej. Spontaniczne przejście PrPC → PrPSc jest rzadkie i prawdopodobnie stanowi efekt mutacji lub wpływu stresu środowiskowego na białko. Wykazano również wpływ wielu różnych białek i czynników molekularnych, wśród których wymienia się: tratwy lipidowe, siarczan heparanu, gangliozyd GM1 i jony miedzi (II) [7].
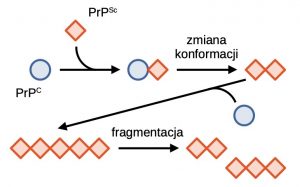
Klasyczny model propagacji zmian prionowych zakłada, że bezpośredni kontakt prion-białko natywne wymusza przejście konformacyjne. Tak powstały agregat stanowi “zarodek” dalszej propagacji zmian prionowych, które postępują z obu końców złogu. Po przekroczeniu pewnej granicznej długości liniowy agregat traci stabilność i ulega pęknięciu. Dwa agregaty potomne stanowią kolejne centra nukleacji złogów – widzimy więc, że mechanizm propagacji prionowej jest samonapędzający się.
Drogi infekcji
Chociaż część chorób prionowych jest wynikiem wystąpienia spontanicznych zmian konformacji białka, to infekcyjny charakter choroby umożliwia zewnątrzpochodne dostarczenie czynnika patogennego. Mechanizm rozprzestrzeniania się prionów po organizmie i ostateczne dotarcie do tkanki nerwowej ściśle zależy od drogi wniknięcia patogenu do organizmu. Wśród szlaków tych szczególną uwagę zwraca się na drogę pokarmową, a dokładniej błonę śluzową jelita [8].
Po dotarciu do błony śluzowej jelita czynnik zakaźny gromadzi się w tkance limfatycznej związanej z jelitem (GALT; ang. gut-associated lymphoid tissue) i dzięki udziałowi komórek układu odpornościowego przekracza barierę krew-jelito. Szczególną funkcję w tym procesie pełnią mechanizmy wychwytu antygenów ze światła jelita: komórki M, makrofagi (komórki te zdają się pełnić ochronną funkcję dzięki aktywności proteolitycznej) i komórki dendrytyczne. Te ostatnie mogą reagować na białka prionowe jak na klasyczne antygeny i transportować je do węzłów chłonnych, skąd czynniki zakaźne mają szansę migrować dalej do tkanki nerwowej [9].
Innym szlakiem migracji białek prionowych po organizmie jest bezpośredni kontakt z tkanką nerwową jelita i przemieszczanie się wzdłuż neuronów. Inwazja ta zachodzi przede wszystkim przy udziale nerwu błędnego, który łączy jelito bezpośrednio z mózgiem. Dokładny mechanizm przemieszania się białek prionowych nie jest znany i wyróżnia się kilka modeli teoretycznych: (I) transmisja złogów białkowych wewnątrz pęcherzyków zewnątrzkomórkowych, które mogą być przekazywane między komórkami w obrębie połączeń synaptycznych; (II) postępujące tworzenie złogów warunkowane oddziaływaniem białek prionowych i fizjologicznych promującym przejście PrPC → PrPSc; (III) poprzez swobodnie przemieszczające się oporne na proteolizę agregaty [10].
Toksyczność agregatów prionowych
Właściwy mechanizm prionowej neurotoksyczności pozostaje nieznany. Nie zostało w pełni zbadane, czy neurotoksyczność jest efektem obecności w tkance form monomerycznych, oligomerycznych czy końcowych włókien amyloidu. Ponieważ jednak obecność fizjologicznej formy PrPC jest niezbędna do aktywacji formy patogennej – wszelkie zmiany prowadzące do nadprodukcji białka PrPC drastycznie zwiększają ryzyko wystąpienia choroby. Wśród zmian tych wyróżnia się chociażby mutacje w obrębie regionu promotora genu PRNP, nadprodukcję czynników transkrypcyjnych i reakcje komórki na nieznane jeszcze bodźce, które wpływają na nadekspresję genu PRNP.
Klasyczne obrazy histopatologiczne chorób prionowych obejmują gąbczaste zmiany tkanki mózgu, utratę neuronów i komórek glejowych. W obrębie zmian udaje się również zaobserwować złogi blaszek amyloidu prionowego.
Sugeruje się, że pierwszym etapem degradacji tkanki mózgowej w chorobie prionowej jest zmniejszenie stabilności synaps. Może to wynikać z preferowanej lokalizacji zarówno białka PrPC jak i PrPSc w obrębie synaps (po stronie błony pre- i postsynaptycznej). Degradacja połączeń synaptycznych skutkuje izolacją neuronów od bodźców i czynników troficznych, co pobudza szlaki proapoptyczne w tych komórkach. Jest to potwierdzone obserwacjami licznych śladów zachodzenia procesu apoptozy, a w tym: fragmentacja DNA i aktywacja kaspazy 3 [4, 11].
Kolejnym istotnym wydarzeniem w trakcie progresji choroby jest stres oksydacyjny, który działa ogólnie niszcząco na komórki ludzkie. Nadmierna produkcja wolnych rodników i reaktywnych form tlenu (ROS; ang. reactive oxygen species) skutkuje uszkodzeniem błon biologicznych, sieciowaniem białek i degradacją materiału genetycznego. W przypadku zaburzeń systemów detoksykacji wolnych rodników i ROS, stres oksydacyjny jest stanem samonapędzającym się, który ostatecznie, prowadząc do uszkodzenia mitochondriów, wzmaga produkcję kolejnych porcji ROS. Na szczególną uwagę zasługuje aktywność antyoksydacyjna fizjologicznej postaci PrPC, która objawia się poprzez wiązanie jonów miedzi (II) w N-końcowej domenie białka. Zmniejszenie ilości aktywnego białka wywołane zmianami prionowymi dodatkowo napędza stres oksydacyjny lub może być jego pierwotną przyczyną. W związku z tym sugeruje się, że niektóre z objawów choroby prionowej mogą być skutkiem utraty neuroprotekcyjnej aktywności białka PrPC, a nie samej obecności złogów prionowych [12].
Jednym z mechanizmów obronnych zaatakowanych prionami komórek są chaperony i proteasomy. Te dwa zespoły czynników odpowiadają za degradację białek nieprawidłowo sfałdowanych i stanowią kluczowy element białkowej homeostazy komórki. Mutacje upośledzające funkcjonowanie tego systemu są jednym z czynników ryzyka wystąpienia chorób prionowych i innych schorzeń neurodegeneracyjnych związanych z obecnością złogów amyloidowych. Prawdopodobnie, często obserwowana w grupach komórek otaczających gąbczaste zmiany mózgu, autofagia jest kolejnym mechanizmem obronnym komórek. Jeden z jej podtypów polega na udziale lizosomów w degradacji wewnątrzkomórkowych złogów białkowych, które są wynikiem ogólno komórkowych zaburzeń (wpływających na homeostazę białkową) lub bezpośrednim skutkiem prionowej polimeryzacji [13].
Inne schorzenia amyloidowe
Wiele chorób neurodegeneracyjnych charakteryzuje się obecnością złogów amyloidowych w tkance OUN. Należą do nich jedne z największych wyzwań współczesnej medycyny: choroba Alzheimera, choroba Parkinsona i stwardnienie zanikowe boczne. Obecnie – mimo odmiennych objawów – coraz częściej opisuje się te choroby wspólnie przywołując zbliżony molekularny mechanizm patogenezy. Możliwość ta sugeruje, że terapia tych chorób również może opierać się na wspólnym mechanizmie [14, 15].
Podsumowanie
Poznanie i dokładniejsze zbadanie chorób prionowych stanowiło jedno z najmniej oczekiwanych wyzwań medycyny. Te patogenne białka o właściwościach zakaźnych z początku wymykały się klasycznym postulatom biomedycyny i biologii chorób zakaźnych, jednak dokładne zbadanie struktur białek prionowych i ich udziału w neurodegeneracji pozwoliło jednoznacznie zaklasyfikować je jako zupełnie nowe czynniki zakaźne. Współczesne badania szeroko pojętych białek o właściwościach prionów pozwoliły nawet odnaleźć ich fizjologiczne odpowiedniki obecne w komórkach drożdżowych, w których uczestniczą w regulacji ekspresji genów.
Rozszerzenie definicji chorób prionowych do formy obejmującej również inne zbliżone choroby neurodegeneracyjne otwiera przed nami zupełnie nowe ścieżki analizowania procesów patogenezy tak kluczowych schorzeń jak choroba Alzheimera, choroba Parkinsona czy stwardnienie zanikowe boczne. Daje to również możliwość opracowania zupełnie nowego i wspólnego podejścia terapeutycznego.
Adrian Macion

Student studiów magisterskich na kierunku Biologia (specjalizacja – Biologia Molekularna) na Uniwersytecie Warszawskim, Prezes Koła Naukowego Biologii Molekularnej na kadencje 2019/20 i 2020/21 i 2021/22, Prezes Koła Naukowego Astrobiologii Voyager na kadencję 2021/22, Założyciel i Redaktor Naczelny Studenckiego Czasopisma Naukowego Eureka!. Obecnie studiuje w Hiszpanii na Universitat Politecnica de Valencia w ramach studiów wymiennych ERASMUS. W ostatnich latach brał również udział w licznych kursach i wymianach organizowanych między innymi przez: Institut Pasteur w Paryżu, niemiecki Ruprecht-Karls-Universität Heidelberg, Università degli Studi di Milano czy Univerzita Karlova w Pradze. Zafascynowany bakteriami, czynnikami i molekularnymi mechanizmami ich patogenezy. Zaangażowany w edukację licealną i akademicką oraz popularyzację nauki. Uwielbia opowiadać o białkach, komórkach i ulubionych historiach Science-Fiction. W wolnych chwilach pisze książki, tatuuje się, maluje obrazy i marzy o badaniu życia na Marsie.
adres korespondencyjny:
Zakład Genetyki Bakterii, Wydział Biologii, Uniwersytet Warszawski
Miecznikowa 1, 02-089 Warszawa
bibliografia
- Liberski PP. Historical overview of prion diseases: a view from afar. Folia Neuropathol. 2012;50(1):1-12.
- Gough KC, Maddison BC. Prion transmission: prion excretion and occurrence in the environment. Prion. 2010;4(4):275-282.
- Sakudo A, Xue G, Kawashita N, Ano Y, Takagi T, Shintani H, Tanaka Y, Onodera T, Ikuta K. Structure of the prion protein and its gene: an analysis using bioinformatics and computer simulation. Curr Protein Pept Sci. 2010 Mar;11(2):166-79.
- Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15(1):34.
- Requena JR, Wille H. The Structure of the Infectious Prion Protein and Its Propagation. Prog Mol Biol Transl Sci. 2017;150:341-359.
- Hirsch TZ, Martin-Lannerée S, Mouillet-Richard S. Functions of the Prion Protein. Prog Mol Biol Transl Sci. 2017;150:1-34.
- Pezza JA, Serio TR. Prion propagation: the role of protein dynamics. Prion. 2007;1(1):36-43.
- Tee BL, Longoria Ibarrola EM, Geschwind MD. Prion Diseases. Neurol Clin. 2018 Nov;36(4):865-897.
- Davies GA, Bryant AR, Reynolds JD, Jirik FR, Sharkey KA. Prion diseases and the gastrointestinal tract. Can J Gastroenterol. 2006;20(1):18-24.
- Moreno JA, Telling GC. Insights into Mechanisms of Transmission and Pathogenesis from Transgenic Mouse Models of Prion Diseases. Methods Mol Biol. 2017;1658:219-252.
- Unterberger U, Voigtländer T. The pathogenic mechanisms of prion diseases. CNS Neurol Disord Drug Targets. 2007 Dec;6(6):424-55.
- Kim JI, Choi SI, Kim NH, Jin JK, Choi EK, Carp RI, Kim YS. Oxidative stress and neurodegeneration in prion diseases. Ann N Y Acad Sci. 2001 Apr;928:182-6.
- Le NTT, Wu B, Harris DA. Prion neurotoxicity. Brain Pathol. 2019;29(2):263-277.
- Ihne S, Morbach C, Sommer C, Geier A, Knop S, Störk S. Amyloidosis-the Diagnosis and Treatment of an Underdiagnosed Disease. Dtsch Arztebl Int. 2020;117(10):159-166.
- Baker KR, Rice L. The amyloidoses: clinical features, diagnosis and treatment. Methodist Debakey Cardiovasc J. 2012;8(3):3-7.